End-to-end framework for simulating the time evolution of a chemical reaction on a fault-tolerant quantum computer
Researchers from PsiQuantum and Stanford have developed a comprehensive state-of-the-art end-to-end framework for the simulation of real-time dynamics of chemical systems on a fault-tolerant quantum computer; this is of interest in modeling the dynamics of chemical reactions, such as those that involve catalysts.
Key Insights
The framework consists of three parts:
(i) constructing an initial state that includes both the chemically active valence electrons and pseudoions (a combination of the chemically inactive electrons and the atomic nucleus)
(ii) evolving the initial state in time with an appropriate, effective Hamiltonian for the chemical system; detailed descriptions of the efficient quantum circuits for the block-encoding of the Hamiltonian are presented
(iii) developing a flexible protocol to identify the interesting chemical species after the time evolution and to extract the information from the quantum state
The entire simulation is performed in first quantization, with each particle (valence electron or pseudoion) represented in a basis of plane waves.
The pseudoions are constructed by combining the chemically inactive electrons of the atom with the atomic nucleus into a single point-charge entity; this approach builds on the well-known concept of pseudopotentials by extending it to a dynamical degree of freedom.
To extract useful chemical information, molecular fingerprints are designed by combining density-functional calculations with machine learning techniques and subsequently validating them through surrogate classical molecular dynamics simulations. These fingerprints are then coherently encoded on a quantum computer for efficient molecular identification via amplitude estimation as the time evolution progresses.
In addition to the newly developed methodology, a detailed calculation of required logical-qubit and logical-operations resources to execute these simulations is provided for a range of interesting chemical systems in three classes of reactions: Ammonia-Boron Trifluoride, Water Gas Shift, and Direct Methane to Methanol with a Pd catalyst; the latter two classes of reactions are particularly interesting for study as catalysts play an important role in them.
The required logical qubits¹ and Toffoli gate counts for a femtosecond of time evolution range from ~800 logical qubits and 10¹¹ Toffoli gates for the reaction between Ammonia and Boron Trifluoride to ~18.8k qubits and 10¹⁴ Toffoli gates for a large simulation of the Direct Methane to Methanol catalytic process.
Context setting: Chemical dynamics and the significance for catalyst-mediated reactions
Understanding the dynamical time evolution of chemical systems at the quantum level remains one of the most complex problems in computational chemistry. Chemical reactions involve intricate processes such as bond formation, bond breaking, charge transfer, and molecular rearrangement, all of which occur at short timescales, often within femtoseconds (10⁻¹⁵ s) to a picosecond (10⁻¹² s). Capturing these processes accurately requires solving the time-dependent Schrödinger equation, which is computationally infeasible for complex molecules using classical computational techniques. Using quantum-computational techniques for elucidating chemical reactions could prove very successful if it is appropriately integrated as part of a larger multi-scale computational framework.
Chemical reactions of particular interest are catalytic reactions, given their industrial significance. Catalysts are widely used in energy production, pharmaceuticals, materials science, and it is estimated that 80% of the world’s manufactured goods are created using catalytic reactions². Moreover, the size of the global industrial catalyst market in 2024 was estimated at ~40 billion USD, and it is expected to grow in the future³ ⁴ ⁵. However, despite their industrial significance, many catalytic mechanisms remain poorly understood due to the limitations of current computational methods.
Most classical approaches rely on the Born-Oppenheimer (BO) approximation, which assumes that electronic motion is instantaneous compared to nuclear motion. This assumption breaks down in catalytic processes where strong electron-nuclear interactions dictate reaction outcomes. Density Functional Theory (DFT), the dominant tool for the calculation of catalytic reactions, relies on the BO approximation as well as heuristic exchange-correlation functionals that are not sufficiently reliable or accurate. Molecular dynamics (MD), another widespread method used for catalysis simulations, captures particle motion but neglects quantum electronic effects, relying on approximate force fields and Newtonian mechanics. Other classical methods are under development, but all involve approximations and trade-offs that limit their wide applicability. In short, classical approaches are insufficient for capturing the full complexity of catalytic reactions.
Quantum computing offers a fundamentally different approach by giving the ability to treat both electrons and nuclei as fully dynamical quantum entities, thereby providing a way to overcome these classical barriers.
Novel research
A key innovation of the study is the pseudoion representation, where an atomic nucleus and its chemically inactive electrons are combined into a single quantum entity. The number of chemically inactive electrons is partially tunable and controlled by the user. This formulation significantly reduces computational overhead by decreasing the number of particles that are considered, while preserving electron-nucleus interactions, making large-scale quantum simulations of chemical systems more feasible. It builds on the technique of pseudopotentials by promoting the pseudopotentials to operators on the full Hilbert space of electrons and pseudoions. The Hilbert space is first constructed separately for each particle using a basis of plane waves (1st-quantization representation), and the total Hilbert space is then constructed as a tensor product of individual Hilbert spaces. The initial state of the joint system of pseudoions and valence electrons that is physically salient can be prepared in a flexible and intuitive manner that is also algorithmically efficient. The quantum circuits for this preparation are explicitly described.
The authors also present optimized quantum circuits for an efficient block-encoding of the Hamiltonian used for the time evolution of the prepared quantum state. Quantum Signal Processing (QSP) is used to implement the time evolution of the quantum state. The block encoding used in this study represents the state of the art in terms of efficiency, reducing previously available overall Quantum Resource Estimates by a factor of ~4 – 6x.
After the time evolution, chemically relevant information is extracted using the newly developed QCI (Quantum Chemical Identification) protocol. The protocol consists of three parts:
Classical computation of fingerprint counting functions for the molecules of interest: fingerprint counting functions are computed classically by training a model to classify molecules of interest based on various features, e.g., bond lengths and angles. For example, molecular dynamics simulations may be used to generate a dataset of molecular configurations that are used for the training of the classifier, with classification based on DFT-computed energies. After training, the fingerprint functions are processed into fingerprint counter functions, ensuring accurate molecular identification while avoiding double-counting or overlap issues
Compilation of the fingerprint counter functions: fingerprint counter functions are compiled for coherent implementation on the quantum computer
Implementation of the fingerprint counter functions: coherently implemented functions are applied on the qubits describing the state of the reaction resulting in the counts for all the chemical species of interest in the reaction. The reaction rate is reconstructed via amplitude estimation
The study explores three reactions to demonstrate the applicability of the framework: the reaction between Ammonia and Boron Trifluoride (NH₃ + BF₃ ⇌ NH₃BF₃), a charge-transfer reaction without a catalyst; Direct Methane-to-Methanol (DMTM) conversion on a Pd-O catalyst complex, which involves complex electron correlations; and the Water-Gas Shift (WGS) reaction (CO + H₂O ⇌ CO₂ + H₂), crucial for hydrogen production and catalyst-surface interactions. The latter two reactions, DMTM and WGS, are catalytic reactions. A detailed quantum resource estimation is provided for the reactions considered. The required logical qubits⁶ and Toffoli gate counts for a femtosecond of time evolution vary by reaction complexity, from ~800 logical qubits and 10¹¹ Toffoli gates for the reaction between Ammonia and Boron Trifluoride to ~18.8k qubits and 10¹⁴ Toffoli gates for a large simulation of the DMTM catalytic process. These results are the first step towards the full quantum study of dynamical chemical systems, with the potential for impact in the catalysis industry.
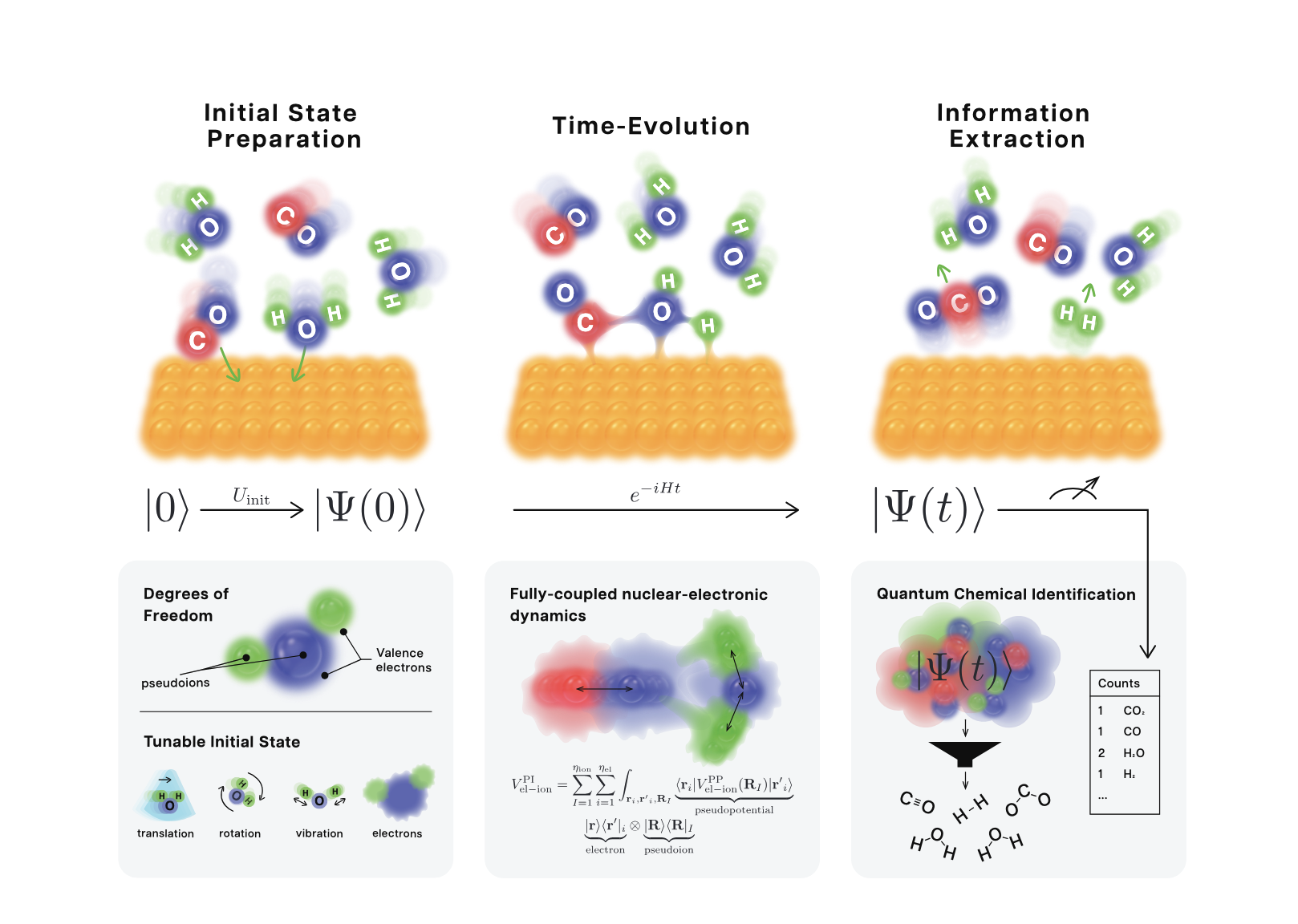
Time-evolution of a catalyst-mediated reaction
Figure 1: Three main steps of the framework for quantum dynamics: (i) Initial state preparation of a physically relevant quantum state of pseudoions and electrons, followed by (ii) time-evolution until the desired final time, and finally (iii) information extraction which includes the identification of chemical species present in the wavefunction.
Conclusion: New paradigm for chemical dynamics and the study of chemical catalysis
This research introduces a comprehensive state-of-the-art quantum approach to chemical dynamics, enabling the direct simulation of real-time electron-nucleus interactions well-beyond the Born-Oppenheimer approximation. The first innovation is the pseudoion-based framework in the plane-wave representation, which compactly encompasses physically relevant effects during the dynamics. The second innovation is a flexible and efficient initial state preparation protocol for molecules and other extended phases. The third innovation is an efficient block-encoding prescription for the interacting pseudoion-electron Hamiltonian. The fourth innovation is the QCI protocol for molecular identification for scalable and efficient identification of reaction products after the time evolution.
The implications of this framework are significant for the study of catalytic reactions and materials because of its ability to address chemical processes, such as bond configuration, with quantum detail that is extremely difficult to reach on a classical computer. Furthermore, this framework expands the possibilities for doing quantum chemistry on fault-tolerant quantum computers beyond static energy calculations, enabling a new class of simulations of dynamics. While large-scale implementations remain hardware-constrained, this research lays a solid foundation for future quantum simulations of catalytic processes and reaction mechanisms.
What’s next?
The authors recognize that there is further work required to advance the framework. Algorithm optimization is one such area, with options to investigate how alternative block-encoding techniques and Hamiltonian representations can reduce quantum resources. Quantum simulations must be compared to state-of-the-art classical methods to understand the limits of approximations and better understand how models represent reality. Eventually, establishing scalable multi-scale simulation workflows for industrial applications that incorporate quantum dynamics simulations could become a way for FTQC to be useful for catalyst design, materials discovery, and cleaner energy solutions.
Essential Terms
Pseudoions: A representation where a nucleus and its chemically inactive electrons are treated as a single point-like quantum entity. This reduces computational complexity while preserving key electron-nuclear interactions, making quantum simulations more practical.
First-Quantized Representation: A representation where electrons and nuclei are treated as individual quantum particles with each having their own basis; the total system space is created as a tensor product of the individual Hilbert spaces.
Block-Encoding: A technique to embed a Hamiltonian into a unitary matrix that can be efficiently simulated on a quantum computer.
Molecular Fingerprints: Suitable combination of relevant chemical information about a molecule. A collection of molecular fingerprints for a reaction of interest can be compiled into counting functions to be applied on the qubits and to determine if those molecules are present in the system. This enables efficient identification of reaction products after simulation.
Quantum Resource Estimation: The process of calculating qubit counts and gate operations required for a run of the quantum algorithm.
Footnotes
¹ excl. auxiliary qubits
² C. Richard Catlow, Matthew Davidson, Christopher Hardacre, and Graham J. Hutchings. Catalysis
making the world a better place. Philosophical Transactions of the Royal Society A: Mathematical,
Physical and Engineering Sciences, 374(2061):20150089 (2016).
³ imarc Group; Catalyst Market Size, Share, Trends and Forecast by Type, Process, Raw Material, Application, and Region, 2025-2033 (2024)
⁴ Precedence research; Catalyst Market Size, Share, and Trends 2024 to 2034 (2024)
⁵ Custom Market Insights; Global Catalyst Market 2025–2034 (2024)
⁶ excl. auxiliary qubits